You are here
Critical Roles of Sensory-Motor Neurotransmission During Normal Development and in Spinal Muscular Atrophy
Speakers

Abstract
Movement is an essential behavior requiring the precise temporal and functional assembly of motor circuits during development. Within spinal motor-circuits, motor neurons act as a bridge between the central and the peripheral nervous system, conveying central commands to the skeletal muscles. Genetic programs specify neuronal circuits assembly during development and perturbations within these programs have deleterious consequences for motor output. During development, the formation and function of motor circuits is thought to be shaped by afferent activity and the output of motor neurons depends on balanced excitatory-inhibitory activity originating from sensory neurons, spinal interneurons and supraspinal pathways. However, the underlying mechanisms of how neurotransmission shapes postsynaptic neuronal output have not been defined. Furthermore, genetic perturbations associated with neurodegenerative diseases may lead to synaptic dysfunction and could serve as initiation points to circuit dysfunction and the demise of normal behavior.
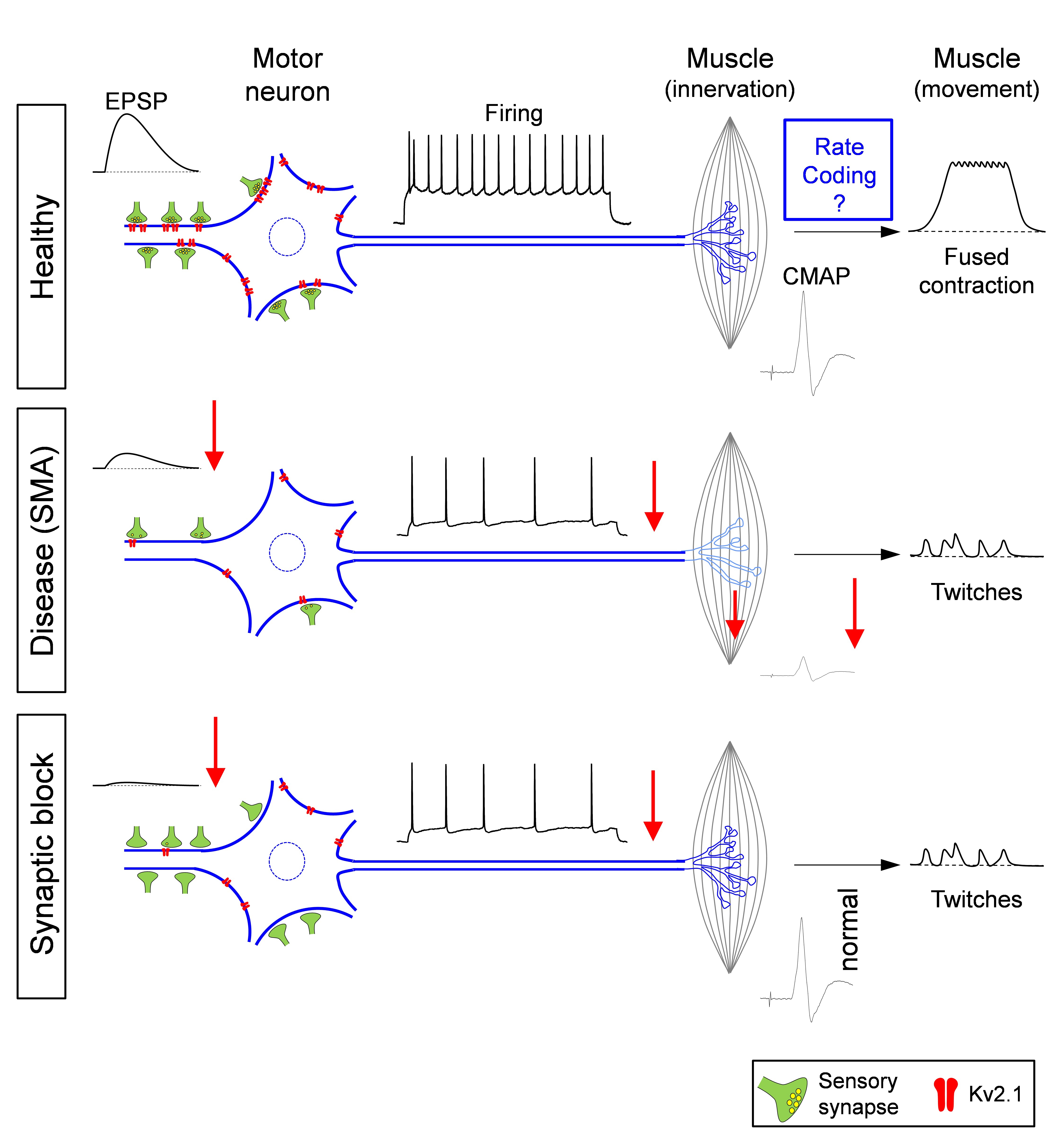
The demise of normal behavior in neurodegenerative diseases is often attributed to selective dysfunction of vulnerable neurons via cell-autonomous mechanisms. Vulnerable neurons however, are embedded in neuronal circuits but the contribution exerted by their synaptic partners to the disease process is largely unknown. We have recently shown show that in a mouse model of spinal muscular atrophy (SMA), an inherited neurodegenerative disease, sensory-motor synaptic dysfunction exerts a powerful influence on the excitability and functional output of motor neurons by reducing their spiking ability in a non-autonomous manner. The synaptic dysfunction is caused by impairment of glutamate release presynaptically from proprioceptive neurons and mediated by a reduction of the potassium channel Kv2.1 on motor neurons, which can be reversed by increasing neuronal network activity pharmacologically. Our results demonstrate a key role of excitatory synaptic drive in shaping the function of motor neurons during development and the contribution of its disruption to neurodegenerative diseases.
The mechanisms responsible for circuit refinement and synapse maintenance are poorly understood. Similarly, the molecular mechanisms by which gene mutations cause dysfunction and elimination of synapses in neurodegenerative diseases that occur during development are unknown. We have recently reported that the complement protein C1q is required for the refinement of sensory-motor circuits during normal development, as well as for synaptic dysfunction and elimination in SMA. C1q tags vulnerable SMA synapses which triggers activation of the classical complement pathway leading to microglia-mediated elimination. Pharmacological inhibition of C1q or depletion of microglia rescues the number and function of synapses, conferring significant behavioral benefit in SMA mice. Thus, the classical complement pathway plays critical roles in the refinement of developing motor circuits, while its aberrant activation contributes to motor neuron disease.