You are here
Amyloid β-Peptide and Brain Oxidative Damage: Focus on the Intersection of the Lipid Peroxidation Product HNE Covalently Bound to Brain Proteins, Glucose Dysmetabolism, and Alzheimer Disease
Speakers

Abstract
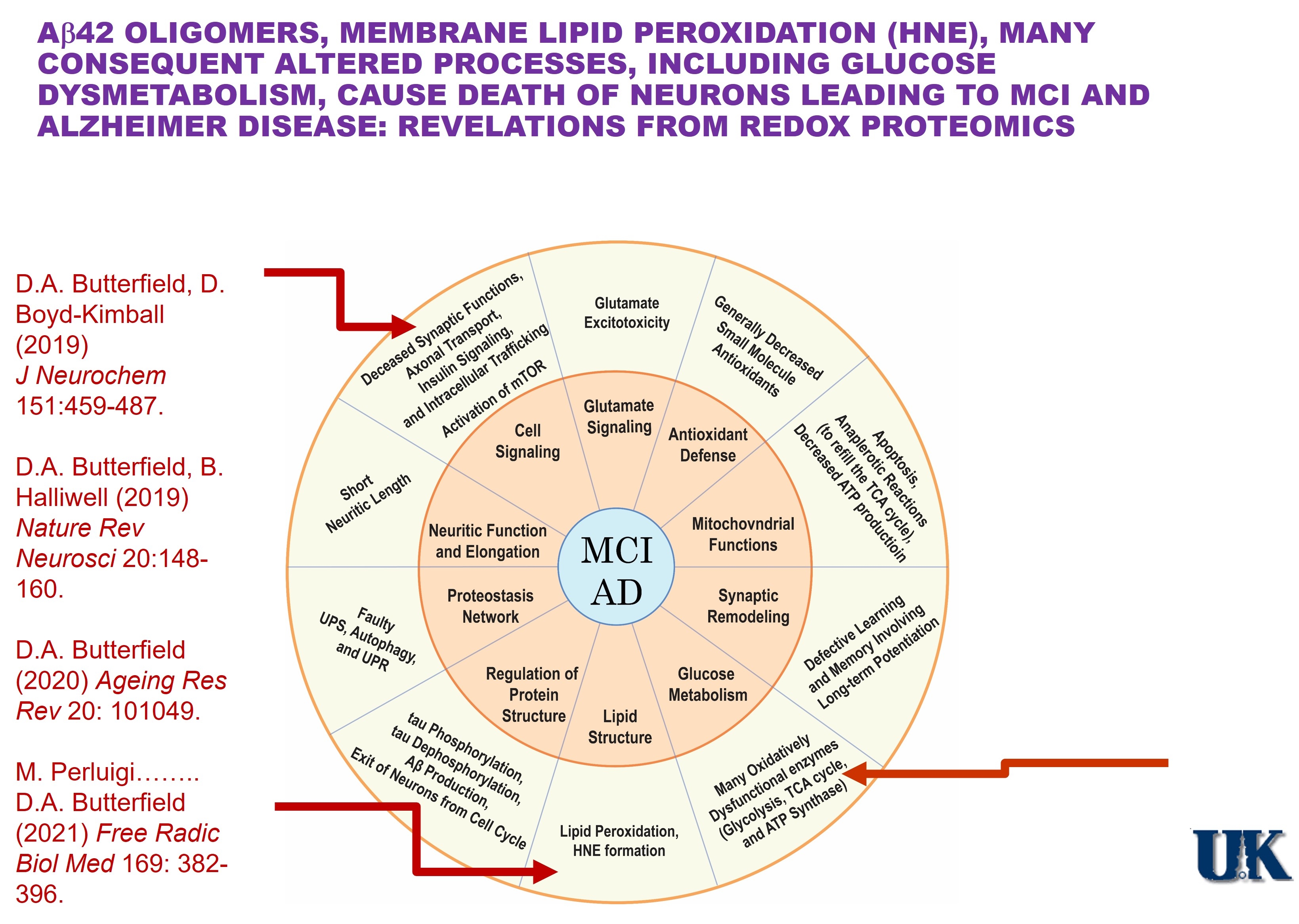
Amyloid β-peptide (Aβ), in oligomeric form (dimers, trimers, quaternary aggregates), was first shown in our laboratory to lead to lipid peroxidation, a major product of which is 4-hydroxynonenal (HNE). A highly reactive molecule, HNE forms covalent Michael adducts with key membrane and cytosolic proteins in neurons, leading to their conformational alterations and loss of function with consequent neuronal death.
Alzheimer disease (AD) is an ever-increasing threat to humanity, and Aβ plays a central role in the development and progression of this dementing disorder. In 1994, our laboratory posited a model to account for Aβ-associated oxidative stress and consequent neuronal death in AD, and to the present date the model has remained viable. Mechanistically, small Aβ oligomers are predicted to enter the lipid bilayer to lead to lipid peroxidation and formation of neurotoxic HNE. Our laboratory pioneered the techniques of redox proteomics leading to the identity of HNE-bound, and consequently dysfunctional, brain proteins, including those associated with glucose metabolism. The resultant diminution of ATP levels leads to loss of neuronal membrane potential due to HNE modification of ion-motive ATPases with resulting inability to maintain calcium gradients. Excess intracellular calcium ion concentration leads to neuronal death of key brain regions associated with cognition, i.e., hippocampus and frontal cortex. Aβ in brain also can initiate processes that cause activation of the mTORC1 pathway with results in inhibition of autophagy and induction of insulin resistance. Our laboratory showed that activation of mTOC1 in brains of persons with mild cognitive impairment (MCI) was associated with insulin resistance, meaning this process is an early occurrence in the progression of AD.
Others using PET and MRI imaging analyses of persons with gene mutations that would lead to familial AD symptoms more than two decades in the future assessed the timeframe and order of symptom presentation. The results showed that the first abnormality observed was deposition of Aβ plaques, followed some years later by glucose dysmetabolism, and lastly by thinning of hippocampal and frontal cortical brain regions due to neuronal death. This scenario precisely mirrors the model of the progression of AD we proposed and provides strongly supportive evidence that Aβ oligomer-associated lipid peroxidation and HNE formation are fundamental processes in the etiology, clinical and pathological characteristics, and progression of AD.
This webinar will outline the evidence obtained in our laboratory to support this conclusion.