You are here
Altered Axonal Actin Dynamics in Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis
Speakers

Abstract
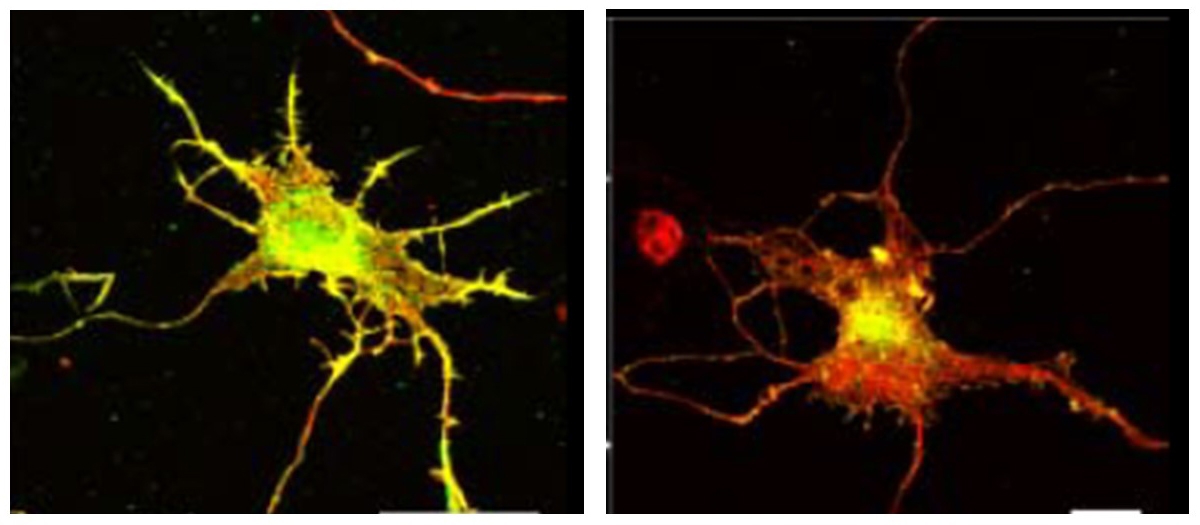
Loss of motor endplates, axonal degeneration and cell death are characteristic pathological features of motoneuron diseases. Distinct gene defects in familial amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) point to different pathophysiological pathways in these two forms of motoneuron disease. These include gain of function mechanisms such as disturbed RNA metabolism and toxicity from dipeptides, but also loss of function of C9ORF72 and SMN that are relevant for synaptic and axonal degeneration. A prominent phenotype of motoneurons from SMA mouse models is reduced axon elongation in the absence of altered motoneuron survival. Smn depletion severely disturbs axonal translocation of a specific subsets of mRNAs. In particular, transcripts associated with axon growth and synaptic activity are reduced, including those coding for α-, ß- and γ-actin. These alterations correlate with disturbed actin dynamics in axons and axon terminals of SMA motoneurons. Similar axonal defects are also observed in C9ORF72 depleted motoneurons. C9ORF72 interacts with cofilin and other actin binding proteins in motoneurons. C9ORF72 loss of function results in enhanced activity of LIM-kinases 1 and 2 (LIMK1/2), acting on downstream small GTPases Arf6 and Rac1 for actin filament dynamic regulation. Thus, common pathological pathways in ALS and SMA exist that could be target for therapy development.